Downregulation of 25-hydroxylase enzyme activity in the liver due to obesity and/or dietary factors which cause obesity are the two newish hypotheses in addition to hypotheses regarding cholecalciferol and/or 25(OH)D being "sequestered" in the excess adipose tissue. Below is my review of the current research, with an additional hypothesis about excess adipocytes consuming 25(OH)D to feed their aberrant vitamin D based intracrine signaling systems.
It is - or should be - well known that people suffering from obesity, in order to attain a given, healthy (such as 50 ng/mL) level of circulating 25-hydroxyvitamin D, need to ingest a quantity of vitamin D3 cholecalciferol per day which is a greater ratio of bodyweight than those who are not suffering from obesity. See the end of this section: https://vitamindstopscovid.info/00-evi/#06-ratios for the statements in Ekwaru et al. 2014 and the 2011 Endocrine Society recommendations.
These statements, when considered in the light of the body weights of people suffering from obesity relative to those who are not, can be interpreted as indicating that a factor of (approximately, and in round figures for simplicity) 1.5 should be applied to the range of ratios of bodyweight(70 to 100 IU/day per kg bodyweight, direct from Afshar et al. 2020) for non-obese people to arrive at the ratios needed for those suffering from obesity. Sunil extended this to a factor of 2.0 for those suffering from morbid obesity, since in the Ekwaru data, the self-assessed category of "obesity" had no separate option for "morbid obesity" and it is reasonable to assume that people suffering from this need a greater amount of vitamin D3 as a ratio of bodyweight to attain the desired 60 ng/mL or more 25(OH)D levels.
As far as I know, the mechanism for this lower than non-obese 25(OH)D level in those suffering from obesity for people taking a given ratio of bodyweight amount of vitamin D3 is generally though of as being due to the excess adipocytes (fat cells, which in obesity are also in places they do not belong) absorbing the 25(OH)D (also potentially vitamin D3) in a way that little or none of it returns to the circulation.
However I found several articles which proposes a separate mechanism concerning the activity of the 25-hydroxylase enzyme. The first of these is based on careful experiments with mice:
Obesity Decreases Hepatic 25-Hydroxylase Activity Causing Low Serum 25-Hydroxyvitamin D
Jeffrey D Roizen, Caela Long, Alex Casella, Lauren O'Lear, Ilana Caplan, Meizan Lai, Issac Sasson, Ravinder Singh, Andrew J Makowski, Rebecca Simmons and Michael A Levine
Journal of Bone and Mineral Research 2019-02-21
https://asbmr.onlinelibrary.wiley.com/doi/10.1002/jbmr.3686
Roizen et al. used a high fat diet to induce obesity in female mice and then measured, with respect to the control group:
- The amount of mRNA which was transcribed for the Cyp2R1
25-hydroxylase enzyme, which is the main enzyme which converts
cholecalciferol to 25(OH)D in the liver. It is also found
elsewhere in the body and these extra-hapatic enzyme molecules
presumably contribute a non-trivial amount of 25(OH)D to the
circulation.
In the liver they found (Fig 1b) ~~40% (my estimate from bar graph) reduction at 21-24 weeks, and similar reductions from 6 weeks.
- With similar levels of circulating cholecalciferol, they
calculated the ratio of circulating 25(OH)D divided by
circulating cholecalciferol and found about 50% (Fig 2a)
reduction in the obese mice.
- By homogenising tissue samples from the liver, they estimated
that the 25 hydroxylation capacity of the liver tissue from the
obese mice was 1/3 (Fig 3) that of the liver tissue from the
mice fed standard chow.
- There was also a matching reduction in the levels of the
25-hydroxylase enzyme in the livers.
- No such reduction in mRNA transcription was found in the liver
for three enzymes which are related to 25(OH)D: Cyp3a11
(Obscure? Does this convert it 25(OH)D to 1,25(OH)2D?),
Cyp24a1 (degrades 25(OH)D to 24,25(OH)D which is broken down and
excreted) and Cyp27a1 (a second and AFAIK less significant
25-hydroxylase enzyme).
- They state (I did not chase the references) that this reduction in Cyp2R1 transcription was caused by:
. . . chronic inflammatory process induced by obesity that involves both innate and acquired immunity, and which is associated with marked increases in circulating cytokines.
Low key sidebar:
Fig 5 shows that in the obese mice force-fed either cholecalciferol or calcifediol (they call it "calcidiol") had differing resultant increases in circulating 25(OH)D. (The numbers on page 1071 are backwards.) The calcifediol raised 25(OH)D levels nearly twice as much as cholecalciferol.
I think they had two significant misunderstandings here. Firstly, they seem to think that IUs can be applied to calcifediol, but this is not the case. An IU applies only to 0.025 ug cholecalciferol. In long-term studies, very approximately, to attain a given rise in circulating 25(OH)D, about 1/14 the amount of calcifediol is needed as vitamin D3. This presumably results from a combination of possibly lower absorption of vitamin D3 and various inefficiencies getting it to the 25-hydroxylase enzyme and then into circulation.
They specified the force-fed amount as "3000 IU / kg bodyweight". I guess they used the same mass of cholecalciferol as calcifediol. It also seems that they expected all the cholecalciferol to be converted in the liver to circulating 25(OH)D. This problems I suggest in this part of their research do not invalidate any other part.
If we make two assumptions:
- The effects observed in this study were largely or entirely
due to obesity in a generalised fashion, rather than to some
significant degree from the particular diet. (See Zhu et al.
2021 below for evidence contrary to this.)
- The relevant biological principles in humans are the same as in mice and the quantitative changes in humans are broadly similar.
then this article gives us good reason to believe that inflammatory or other obesity-driven processes (or dietary factors which drive obesity) directly diminish the ability of the liver (and perhaps other tissues) to hydroxylate cholecalciferol to 25(OH)D and that this plays a significant role in the need for greater ratios of bodyweight to be used for people suffering from obesity when recommending optimal vitamin D3 intakes.
A second article is:
Obesity Represses CYP2R1, the Vitamin D 25-Hydroxylase, in the Liver and Extrahepatic Tissues
Mahmoud-Sobhy Elkhwanky, Outi Kummu, Terhi T Piltonen, Johanna Laru, Laure Morin-Papunen, Maija Mutikainen, Pasi Tavi and Jukka Hakkola
Journal of Bone and Mineral Research Plus 2020-07-20
https://asbmr.onlinelibrary.wiley.com/doi/full/10.1002/jbm4.10397
This builds on work with male mice by the same team: Aatsaki et al. 2019 which showed that:
Cyp2R1 and its catalytic activity vitamin D 25-hydroxylation are suppressed in the liver during fasting and in both type 1 and type 2 diabetes mouse models. Mechanistically, we demonstrate involvement of at least two molecular pathways: the peroxisome proliferator–activated receptor gcoactivator 1-a(PGC-1a)/estrogen-related receptor a (ERRa) axis and the glucocorticoid receptor (GR). Furthermore, CYP24A1 is induced during fasting under the control of PGC-1a. Altogether, these results indicate that energy metabolism–regulating factors control vitamin D metabolism and establish repression of vitamin D bioactivation as an important, novel mechanism inducing vitamin D deficiency in diabetes.
Elkhwanky et al. cite Roizen et al. 2019 and their own work as noted above. They conducted further work with mice and attempted to research similar processes in humans. They found that in both male and female obese mice:
- Transcription of Cyp2R1 mRNA in the liver was suppressed by
(my estimate of Fig 3B) 80 to 90%, which is a drastic change.
- Liver levels of the Cyp2R1 enzyme were reduced about 65% (my
estimate of Fig 3C).
- There were a variety of changes in Cyp2R1 mRNA levels in a variety of tissues. The details are too complex to me to make much sense of or mention here. Likewise levels of the Vitamin D receptor in various tissues.
They also investigated the effect of fasting on Cyp2R1 mRNA levels. I was surprised to read:
Fasting and obesity involve activation of partially similar hormonal and signaling mechanisms.
They also found that:
Fasting and activation of glucocorticoid receptor represses Cyp2R1 in the kidney
but I think this is peripheral to my focus on the Cyp2R1 mRNA and
its 25-hydroxylase enzyme in the liver.
Their human research was unable to directly measure Cyp2R1 mRNA or 25-hydroxylase enzyme levels or activity in the liver, but they did research expression of the mRNA of this and other enzymes in subcutaneous abdominal adipose tissue samples taken from four morbidly obese women before Roux-en-Y gastric bypass surgery and another set of samples taken 11 to 19 months after surgery, during which time they had lost between 20 and 42% of their pre-operative weight.
They regard Cyp2R1 expression as being rather similar in many tissues, so these subcutaneous samples may indirectly indicate what is occurring in the human liver. They found an average 1.5 factor (50% increase) in the post-operative mRNA expression levels, with similar increases for all four subjects. This could be used to infer similar changes in the human liver, as can the observations in mouse liver.
Since it would be unethical to induce obesity in human subjects
and probably to take liver biopsies in subjects whose obesity was
reduced significantly by surgery or other means we may not be able
to confirm more directly that obesity reduced 25-hydroxylase
enzyme activity in the human liver, which is the central
question. Likewise, as Zhu et al. below suggest, regarding
dietary factors which drive obesity, rather than obesity itself.
However, I regard the above research as indicating that this effect can reasonably be expected to occur in the human liver. It would take a significant biological difference between humans and mice, which is not known to exist, for this not to be the case.
Elkhwanky et al. suggest that in humans, the role of the liver in creating circulating 25(OH)D may not be as great as is commonly assumed. I have heard statements to this effect and have not yet tried to understand this better. They wrote:
Interestingly, according to the human tissue atlas, Cyp2R1 appears to be expressed rather ubiquitously in most tissues, and the liver expression is not particularly high among human tissues.
From their Supplementary Materials PDF: https://asbmr.onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fjbm4.10397&file=jbm410397-sup-0001-SupInfo.pdf the graph does indeed suggest to me, as others have told me, that skin conversion of cholecalciferol may be significant:
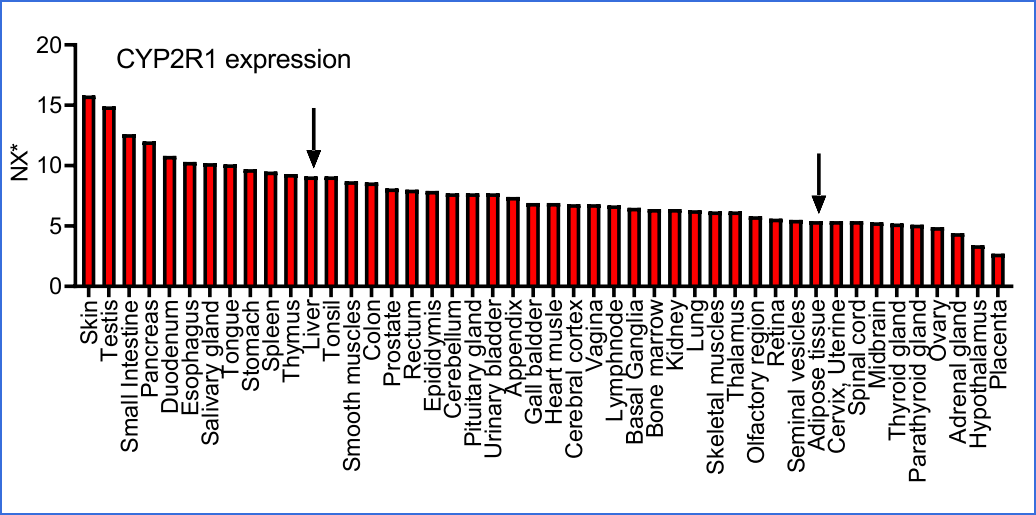
In terms of volume, I guess the adipose tissue, skin, liver and
small intestine are the most significant tissues which might
affect circulating 25(OH)D levels, though skin would be more
important for natural UV-B synthesised cholecalciferol than that
which is obtained orally.
Elkhwanky et al.discuss other mechanisms which have been proposed for low circulating 25(OH)D levels in people suffering from obesity. Lack of sun exposure is one, but I am concerned about the amount of vitamin D3 to supplement orally to most likely attain a 50 ng/mL or more circulating 25(OH)D level, without the need for medical involvement or 25(OH)D testing. The other two hypotheses mentioned are:
- Trapping (sequestration?) of vitamin D3 cholecalciferol in
excess adipose tissue, due to its hydrophobicity. I think this
implies that the vitamin D3 is degraded there or, if
hydroxylated to 25(OH)D there for that 25(OH)D to be consumed
there (as feedstock for intracrine / paracrine signaling) and/or
degraded there by the 24-hydroxylase enzyme.
- Volumetric dilution of vitamin D3 cholecalciferol due to large body size.
The first could explain the apparent need for vitamin D3 supplemental intakes needing to be based on higher ratios of bodyweight in order to attain 50 ng/mL or more 25(OH)D in people suffering from obesity, but the second could not.
There is also another hypothesis, like the first, which I may have read somewhere - or not - that excess adipose tissue absorbs and consumes circulating 25(OH)D. It seems that few vitamin D researchers are aware of vitamin D based intracrine and paracrine signaling, best known in some types of immune cells: https://vitamindstopscovid.info/00-evi/#02-compounds . I recall reading (no refs handy just now) that in obesity, these excess adipocytes behave like immune cells and emit pro-inflammatory cytokines. If so, they may be using vitamin D based intracrine signaling to do this. If so, this would consume 25(OH)D, and the intracellularly produced 1,25-dihydroxyvitamin D would be consumed locally by enzymes (such as 24-hydroxylase) quite rapidly, as is (as best I understand it) normally the case with intracrine signaling: to make the mRNA transcription and so cellular behavioral changes respond rapidly to the intracrine signaling system being turned on and off according to the circumstances.
A recent article:
High Fat Diet and High Cholesterol Diet Reduce Hepatic Vitamin D-25-Hydroxylase Expression and Serum 25-Hydroxyvitamin D3 Level through Elevating Circulating Cholesterol, Glucose, and Insulin Levels
Tengfei Zhu, Jingyu Zhao, Shu Zhuo, Zhimin Hu, Shuyu Ouyang, Wunier, Shuting Yu, Yan Chen, Yu Li and Yingying Le
Molecular Nutrition and Food Research 2021-08-27
https://onlinelibrary.wiley.com/doi/full/10.1002/mnfr.202100220
pursued further research with mice. This broadly corroborates the observations about obesity and 25-hydroxylase activity mentioned above, but, crucially, attributes the changes to 25-hydroxylase activity to several aspects of the diet which normally results in obesity, rather than to the obesity itself:
Low circulating 25-hydroxyvitamin D (25(OH)D) levels associate with obesity, diabetes, and hyperlipidemia, but the underlying mechanisms remain uncertain. As energy-dense diet contributes to these disorders, this study investigates whether diet could impair vitamin D metabolism.
Methods and Results: Compared with control chow-fed mice, high fat diet (HFD)-fed mice show lower serum 25(OH)D3 and 1,25(OH)2D3 levels, lower hepatic vitamin D 25-hydroxylase Cyp2R1 expression but comparable renal vitamin D metabolic enzymes expression. Time course studies show that after HFD feeding, the serum concentrations of cholesterol, triglycerides, fatty acids, glucose, and insulin elevate sequentially and before the reduction of hepatic Cyp2R1 expression and serum 25(OH)D3 levels. Hepatic Cyp2R1 expression also reduces after consuming high fat and high sucrose diet. After high cholesterol diet feeding, serum total cholesterol rises and hepatic Cyp2R1 expression decreases ahead of the reduction of serum 25(OH)D3.
In-vitro studies demonstrate that high concentrations of cholesterol, glucose, and insulin significantly inhibit Cyp2R1 expression in primary murine hepatocytes. Further studies show that dietary restriction in HFD-fed mice ameliorates hypercholesterolemia, hyperglycemia, and hypertriglyceridemia, and elevates hepatic Cyp2R1 expression and serum 25(OH)D3 level.
Conclusion: These findings suggest that diet-induced elevation of circulating cholesterol, glucose, and insulin reduces serum 25(OH)D3 level through suppressing hepatic Cyp2R1 expression.
My current thinking is to summarise the situation like this:
The reasons for people suffering from obesity needing a higher ratio of bodyweight quantity of supplemental vitamin D3 cholecalciferol than non-obese people, in order to obtain 50 ng/mL circulating 25(OH)D, are not known with any certainty.
One hypothesis is that vitamin D3 is absorbed and consumed - such as by 24-hydroxylation - by the excess adipose tissue. Another is that the same process happens to 25(OH)D, with it perhaps being consumed in autocrine signaling in those cells - as they are known to produce pro-inflammatory cytokines.
A second hypothesis has gained experimental support in mice. These experiments cannot be extended to humans, but it would not be surprising if the same processes occurred in humans. The expression of the Cyp2R1 gene in mouse livers and other tissues has been shown by two groups to be significantly downregulated in obesity. (Cite Elkhwanky.) This has been shown to reduce the amount of 25-hydroxylase enzyme in the liver, to reduce the 25-hydroxylase activity of homogenised liver samples and to reduce the level of circulating 25(OH)D significantly. Similar outcomes have been reported by Zhu et al. 2021, but due to high fat and high sugar dietary intakes which in part cause obesity, rather than due to obesity itself.
Whatever the cause for this need for increased vitamin D3 supplemental intake as a function of bodyweight, it is important that people suffering from obesity, and especially morbid obesity, are advised to use higher ratios of bodyweight to calculate desirable vitamin D3 intakes. They face numerous health challenges, their condition is in part an inflammatory disorder and higher 25(OH)D levels are likely to help reduce the immune system dysregulation which causes this pervasive inflammatory state.